
Come si manifesta la fibrosi cistica – Sintomi
Sintomi poco dopo la nascita
La fibrosi cistica è una malattia genetica che interessa diversi organi. Per questo già poco dopo la nascita si avvertono diversi sintomi:
- non viene espulso il meconio, le prime feci del neonato, il che comporta un blocco intestinale (ileo da meconio);
- i bambini hanno un «sapore salato» sulla pelle;
- i neonati rimangono sottopeso, soffrono di flatulenza e dolori addominali, le feci sono grigiastre e viscose;
- i bambini iniziano a soffrire molto presto di tosse cronica e disturbi respiratori.
Muco denso nelle vie respiratorie
La fibrosi cistica colpisce le ghiandole secretorie portando alla formazione di un muco molto denso che impedisce il normale ciclo di pulizia nelle vie respiratorie e l’espulsione delle impurità e batteri. Ne conseguono ricorrenti infezioni delle vie respiratorie e tosse cronica. Con il progredire della malattia si arriva alla distruzione dei tessuti polmonari, con conseguente dispnea e mancanza di ossigeno.
Digestione compromessa
Il muco denso che colpisce le persone affette da fibrosi cistica ostruisce i dotti del pancreas, impedendo agli enzimi della digestione di affluire all’intestino. La digestione risulta così incompleta e, venendo a mancare importanti sostanze nutritive, i bambini tendono a non svilupparsi come dovrebbero e gli adulti a rimanere sottopeso.
Negli adulti possono manifestarsi anche sintomi e patologie concomitanti quali: infiammazioni del fegato e della cistifellea, diabete e osteoporosi.
Cause e possibilità di trattamento
Cosa provoca fibrosi cistica – Cause
La fibrosi cistica insorge a causa di un difetto nel patrimonio genetico che i genitori trasmettono ai figli. Nel 1989 gli scienziati riuscirono a localizzarlo nel numero 7 dei 23 cromosomi complessivi. In quel punto nei soggetti affetti da fibrosi cistica è memorizzata un’informazione errata, che impedisce la produzione corretta di un’importante proteina, la CFTR (Cystic Fibrosis Transmembrane Conductance Regulator). Nelle persone sane, questa proteina garantisce che il muco e le secrezioni siano sufficientemente fluide. Quando è difettosa, la produzione di liquidi e sali nell’organismo è completamente compromessa: i fluidi corporei sono densi e collosi e provocano l’ostruzione di organi vitali quali il pancreas, l’intestino e soprattutto i polmoni.
Genitori sani, bambini malati
In Svizzera, quasi una persona su 20 è portatrice del gene difettoso sul cromosoma numero 7. Tuttavia, un neonato su 3300 si ammala di fibrosi cistica. Come mai? Un bambino si ammala di fibrosi cistica quando entrambi i genitori sono portatori di un cromosoma difettoso ed entrambi trasmettono il gene difettoso. Se il bambino eredita il gene difettoso da un unico genitore, non si ammalerà di fibrosi cistica, ma potrà trasmettere la malattia per via ereditaria (portatore sano).
Accertare la fibrosi cistica – Diagnosi
Screening neonatale
Dal 2011 tutti i neonati vengono sottoposti al test della fibrosi cistica. Il quarto giorno dopo la nascita, vengono prelevate delle gocce di sangue dal tallone, con cui si effettua lo screening di dieci patologie ormonali e del metabolismo, tra cui la fibrosi cistica.
Test del sudore
In caso di risultato sospetto, un test del sudore può confermare la diagnosi. Nei neonati affetti da fibrosi cistica il tenore di sale nel sudore è più alto. È un test indolore, che nel 98% dei casi risulta positivo.
Test genetico
Il test genetico prevede il prelievo di una goccia di sangue o di un tampone salivare per individuare il difetto genetico.
Diagnosi prenatale
Già nei primi mesi di gravidanza è possibile stabilire se il neonato è affetto da fibrosi cistica. Tra l’ottava e la dodicesima settimana di gravidanza viene effettuato il prelievo di un campione di tessuto dalla placenta (villocentesi) oppure di una piccola quantità di liquido amniotico (amniocentesi) tra la quattordicesima e la sedicesima settimana.
Aspettativa di vita più alta – Trattamento
Tutte le forme di trattamento mirano al mantenimento, il più a lungo possibile, della funzione degli organi colpiti. Nuove terapie permettono oggi di allungare notevolmente l’aspettativa di vita e migliorare la qualità della vita dei pazienti. I neonati a cui è stata diagnosticata la malattia attraverso lo screening neonatale hanno buone possibilità di raggiungere l’età pensionabile.
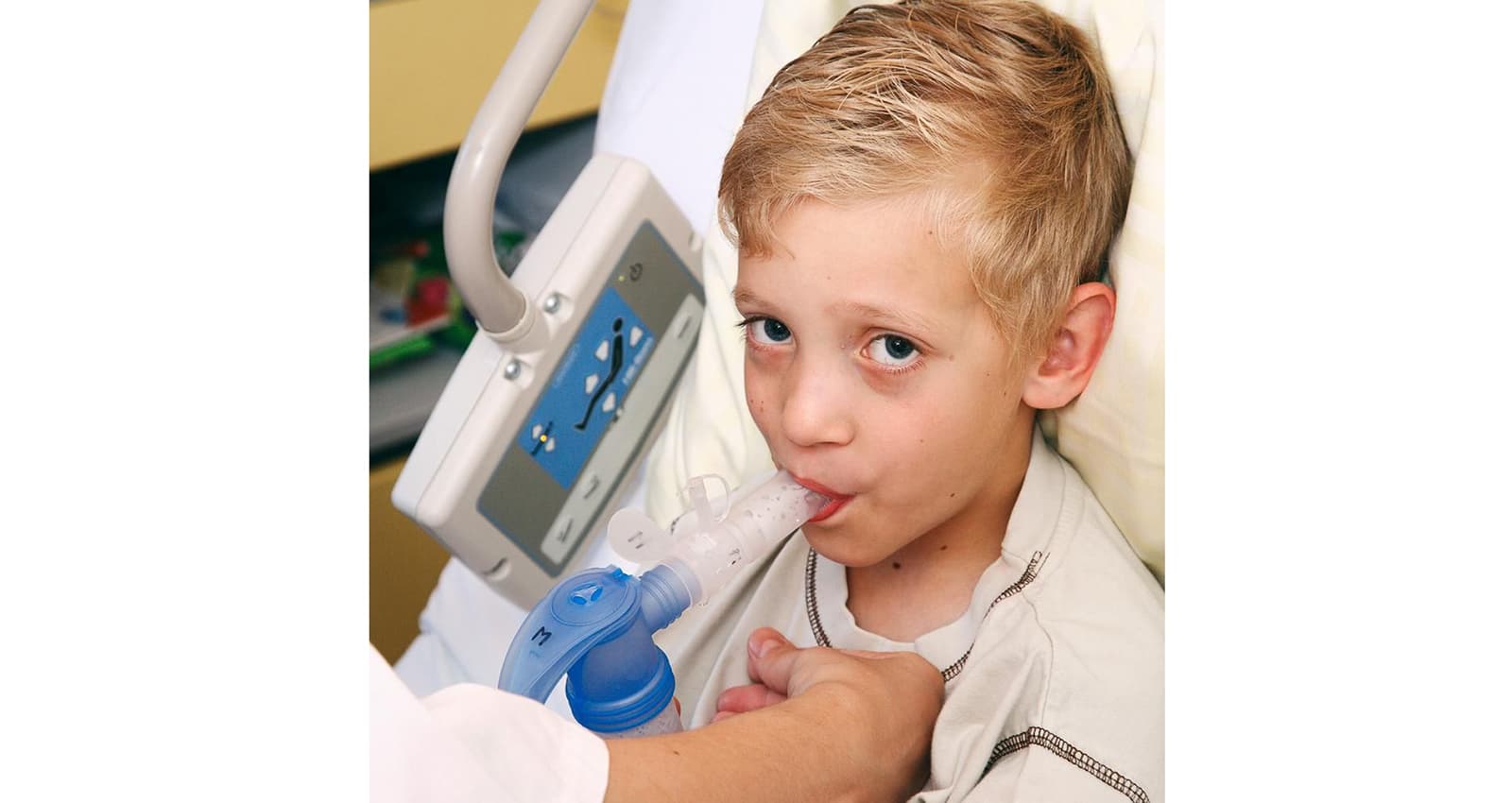
Fisioterapia respiratoria per migliorare la funzione polmonare
I trattamenti fisioterapici accompagnano le persone affette da FC per tutta la vita. L'offerta di assistenza fisioterapica comprende in prima linea la fisioterapia inalatoria e respiratoria, da integrare con esercizi di forza e resistenza e tecniche di rilassamento. Con la terapia respiratoria è possibile migliorare la funzione polmonare grazie a speciali esercizi che permettono di sciogliere ed espellere il muco denso dai bronchi. Questo agevola la respirazione e riduce il rischio di accumulo di agenti patogeni.
Antibiotici contro le infezioni batteriche
Le ricorrenti infezioni delle vie respiratorie provocano la lenta distruzione dei polmoni. Spesso l’infezione può essere attribuita a ceppi di batteri che gli antibiotici non riescono a tenere sotto controllo. L’uso frequente di antibiotici rende però più soggetti a fenomeni allergici o di resistenza agli stessi.
Ripristinare la corretta funzione del canale del cloro
Nuovi medicamenti, i cosiddetti modulatori CFTR (Cystic Fibrosis Transmembrane Conductance Regulator) hanno permesso in tempi recenti di migliorare o addirittura ripristinare la funzione della proteina CFTR (canale del cloro). Ciò consente di diminuire o anche interrompere le terapie sintomatiche.
Più calorie e enzimi digestivi
A causa delle infezioni e dell’insufficiente funzionalità del pancreas, le persone colpite da fibrosi cistica soffrono spesso di sottoalimentazione. Hanno perciò bisogno di una quantità maggiore di calorie e vitamine rispetto alle persone sane. Affinché l’organismo assimili le sostanze nutritive, prima e durante i pasti i pazienti devono assumere anche dei preparati a base di enzimi che li aiutino ad assimilare le sostanze nutritive.
Trapianto dei polmoni
Se nonostante l’intensa terapia, il degrado dei polmoni continua a progredire, il trapianto di polmone può essere una possibilità da prendere in considerazione quando, ad esempio, l’aspettativa di vita del paziente è inferiore a due anni o la funzione dei polmoni è fortemente limitata oppure ancora in caso di un continuo susseguirsi di gravi infezioni. La decisione a favore o contro un trapianto dipende da numerosi fattori.
In caso di fibrosi cistica si rende necessario un trapianto di entrambi i polmoni. I problemi principali dopo un intervento di questo tipo sono il sopraggiungere di infezioni e il rigetto del nuovo organo.

Corsi & Offerte per le persone colpite


Avete delle Domande? Siamo a vostra disposizione!
Le Leghe polmonari cantonali offrono un servizio completo di consulenza e assistenza. L’obiettivo è aiutare le persone colpite a vivere il più possibile senza disturbi. Se avete dubbi o domande, rivolgetevi alla Lega polmonare cantonale più vicina.
